One of the most common drugs we encounter in the field are various forms of anticoagulant and antiplatelet medications. These are relevant to our care both in their therapeutic role as well as in their adverse reactions and potential for harm.
Unfortunately, coagulation is a miserably complex process, and it has to be understood at least generally in order to understand these drugs. In the hope of making this less confusing, rather than throw a wall of text at you, the worker gnomes at EMS Basics have put together an illustrated video. View this, then read on — the drugs won’t make any sense if you don’t start with the physiology.
This form of teaching is a new frontier here, so any input or feedback is welcome. Due to both personal and technical failings, it didn’t turn out exactly how I’d hoped, but hopefully things will continue to improve in the future.
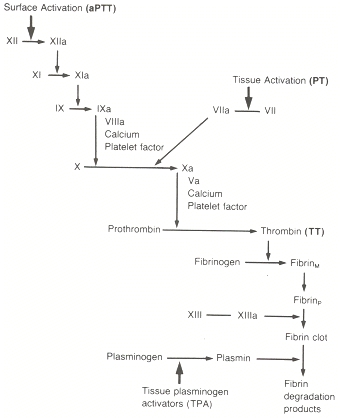
Now that we understand the process, we should talk about the drugs.
There are two major categories here: anticoagulants and antiplatelets. Antiplatelet drugs inhibit the initial step of platelet aggregation and adhesion, where they collect at the wound site in activated form and create a loose plug. Anticoagulants have no effect on this, but instead interfere with the production of fibrin, and therefore prevent a solid clot from growing.
As a general rule, the anticoagulants are rather more clinically significant, as far as their effects on bleeding.
Anticoagulants
First off, to be clear: tPA (tissue plasminogen activator) is not an anticoagulant of any shade. It is a thrombolytic; it attacks and degrades existing clots, dissolving their fibrin bonds. It has no role as a protective agent, and would be far too hazardous in such a role anyway; even its emergency use for acute events like ischemic stroke always requires careful weighing of benefit vs. risk — because the risks are significant.
With that said, there are two main anticoagulants we see frequently in the field.
Warfarin (Coumadin)
Coumadin is an old drug with an interesting backstory; one of its original uses was for rat poison. It’s given orally.
Nowadays, it’s mainly used for chronic anticoagulation of patients at high risk for embolic events. For instance, if you’re in atrial fibrillation at baseline, the blood in your atria isn’t being pumped downstream effectively, and tends to pool. We saw that brisk movement of the blood is one of the main ways we prevent clotting; A-fib is therefore a risk factor for hazardous clots. So when possible, these patients are covered by Coumadin or similar drugs.
The mechanism is interesting. Recall that for the activation of several factors, primarily in the extrinsic and common pathways (including thrombin and Xa), Vitamin K needs to be present. (For some factors, Vitamin K is also needed for the initial production of their inactive forms.) The process looks like this: in order for the factors to be activated, a second background process must also occur, where Vitamin K is changed into a form called Vitamin K epoxide. Once this is done, Vitamin K epoxide can be cycled back into Vitamin K, allowing it to be reused again for the next activation.
Coumadin prevents this second step. It allows the inital activation and conversion, but it blocks Vitamin K epoxide from being recycled to Vitamin K. So over time, as you use up available Vitamin K, it doesn’t get replaced, and you end up with less and less of it available. Less available Vitamin K means less activation of thrombin and its precursors, which means less fibrin, which means less clotting.
Obviously this process takes time. Since Coumadin has no effect on the active factors already present, if we start you on Coumadin today, it won’t have any effect for several days. We need to wait for currently circulating factors to degrade. So for newly anticoagulated patients, a more fast-acting drug is usually used to cover this loading period; heparin is common.
Other than its widespread use, warfarin is also famous for frequent misdosing. It has a narrow therapeutic index, where it’s very easy to give too much or too little, and depending on diet and other drugs, the appropriate dose can change daily. It therefore requires regular monitoring of the patient’s actual anticoagulation, which is done through a test called the prothrombin time (PT). This is a lab test that measures clotting time with an emphasis on the extrinsic and common pathways, and gives a result in seconds. Due to different PT tests available, a standardized result has been devised called the INR (or International Normalized Ratio). This is essentially a ratio of your clotting time over the standard clotting time; a normal result is therefore close to 1.0. Obviously, anticoagulated patients should have a longer clotting time, so 2.0–3.0 is more typical. Much higher than this puts one at high risk of bleeding — into the GI tract, into the lungs, into the nose and mouth, and if trauma occurs, the chance of significant bleeding is magnified. A too-low INR, of course, simply removes the benefits of protective anticoagulation.
In the event of overdoses that need reversal, patients can receive supplemental Vitamin K, as well as plasma (or concentrates) to replace the missing factors directly.
Heparin
Heparin is another old drug. It’s actually a biological substance naturally present in the blood, one of the body’s own anticoagulants, and when extracted for pharmacological use it’s derived from sources like pig intestines. Lovely. You can’t take it orally, so as a rule it’s given by IV.
Compared to warfarin, heparin has a more direct mechanism. Recall that one of the antagonistic factors that works to deactivate thrombin (as well as a few other factors) is antithrombin. Heparin, when taken in therapeutic doses, multiplies the effects of antithrombin by several thousand times. It therefore deactivates far more factors, which are then unable to produce fibrin. Thrombin and factor Xa are two of the factors most affected.
You can already imagine that heparin will probably work much faster than Coumadin. Aside from being given intravenously, it’s not simply stopping the influx of new Vitamin K and waiting for the old factors to degrade; it’s actually going in and deactivating them directly. In fact, heparin takes effect within half an hour or so. However, its half-life is short, so it’s often given as a continuous drip. Obviously, its usage is typically for acute events, such as acute coronary syndromes, or the bridging to Coumadin we mentioned.
However, there is another version of heparin that’s available. To briefly describe the chemical structure of heparin, it’s a polysaccharide, or a repeating chain. When we cook this stuff from pig parts, we end up with a collection of heparin chains in widely varying lengths. The problem is that only chains of a relatively long length will deactivate thrombin. So depending on the actual size of our heparin molecules, unaltered heparin — known as unfractionated heparin — can be fairly unpredictable in its effectiveness as an anticoagulant.
Even very short chains, however, will deactivate factor Xa, and since Xa is a necessary precursor for thrombin, this has the same effect. So if we can produce an artificial product that only includes short heparin chains, then it will mostly affect Xa rather than thrombin, and its effects will be more predictable. This is called low molecular weight heparin, and it has several advantages. It’s easier to manage, it requires less close monitoring, and it has a longer half-life. In fact, it can be given once a day by subcutaneous injection; for instance, post-operative patients can be taught to inject themselves and sent home with the ability to manage their own anticoagulation. Most of these LMWHs end in -arin: enoxaparin (Lovenox), dalteparin (Fragmin), and tinzaparin (Innohep) are common. Fondaparinux (Arixtra) is also used; although technically not a LMWH, it’s very similar in all respects.
Heparin can be monitored by testing the partial thromboplastin time (PTT), which focuses on the intrinsic and common pathways. LWMH can, if necessary, be monitored by testing levels of factor Xa. Overdose leads to bleeding complications, and in a few cases heparin can induce a disorder called heparin-induced thrombocytopenia (HIT), causing a paradoxically elevated chance of clotting. Super-therapeutic levels can be reversed by protamine sulfate, which binds to heparin and prevents its utilization.
Dabigatran (Pradaxa)
A few brief words on this relatively new drug, only made available over the past year or so.
Dabigatran is an anticoagulant from a wholly different class known as direct thrombin inhibitors. Unlike the somewhat roundabout pathways of warfarin and heparin, these drugs inhibit thrombin directly, and may therefore be somewhat more predictable and easily managed.
In the case of dabigatran, it’s being marketed as a replacement for Coumadin. Although supposedly just as effective for chronic anticoagulation, its claim to fame is that it requires no monitoring of INR, which would be a huge burden lifted from patients and caregivers.
Still very new, it remains to be seen how widely it will be adopted. The main concerns about it are: 1. Cost, and 2. Reversal. Unlike warfarin, which in the case of hazardous events (the proverbial bonk-to-the-head with an epidural bleed) can be readily reversed by Vitamin K and fresh frozen plasma, there is no easy or clear method of reversing dabigatran. Some ideas are out there, but clinical experience remains scarce at this point. In any case, this drug isn’t too common yet, but you may start to see it more often.
Antiplatelets
Aspirin
Aspirin is probably in your medicine cabinet somewhere. It has widespread uses from analgesia to antipyretic effects, but also plays a role in platelet adhesion. It’s taken orally, although IV aspirin does exist, and is used both for chronic risk-reduction and acute treatment of coronary syndromes. This stuff is good enough that nearly everybody you know with wrinkles on their face probably takes it every day.
As platelets are activated and degranulate, one of the chemicals they release is thromboxane A2. It has several effects, including vasoconstriction of the immediate area and stimulating further platelet activation. However, it also promotes platelet adhesion by a pretty neat mechanism.
Remember fibrinogen? The inactive precursor of fibrin? Unlike some of the other inactive factors, this one has its own chance to be the star of the show. Fibrinogen can form a bond between activated platelets, attaching at their glycoprotein IIB/IIIA receptors and creating a link. This isn’t anywhere near as strong as a fibrin bond, but it’s enough to make platelets stick together and clump. Thromboxane activates glycoprotein IIB/IIIA receptors and allows the formation of these fibrinogen bridges.
Aspirin inhibits thromboxane release. Fewer fibrinogen bonds are formed, and less platelets adhere. Coagulation itself proceeds unimpeded, but there are fewer platelets in the clot to be married by fibrin.
Due to the widespread effects of aspirin, overdose is a complex subject. Altered mental status, neurological and cardiovascular signs, sensory disturbances (blurred vision or ringing of the ears), and GI problems are all possible. However, there are typically no obvious bleeding abnormalities. Treatment of acute toxicity can include attempts to limit the dosage (such as gastric lavage and activated charcoal), bicarb, supportive care, and if necessary hemodialysis.
Glycoprotein IIB/IIIA inhibitors
This mouthful of a name is another class of drugs from the antiplatelet family. They’re typically not used chronically like aspirin; one reason is because they’re given intravenously, with oral forms rarely seen. (Another reason is because they’re simply stronger drugs). We see these most often used during and after known coronary “events,” such as a STEMI, NSTEMI, or a coronary catheterization, at which times they can help prevent reocclusions.
Their mechanism is similar to aspirin. As we saw, fibrinogen binding to glycoprotein IIB/IIIA receptors helps bind together platelets and allows them to adhere and aggregate. GBIIB/IIIA inhibitors block these receptors by competitive binding, and hence prevent the fibrinogen bonds.
We rarely see these in the field, but common ones include: abciximab (ReoPro), eptifibatide (Integrilin), and tirofiban (Aggrastat). Adverse effects mainly involve bleeding.
Thienopyridines
Although there are a few drugs in this class, by far the most common is clopidogrel (Plavix). Think of these as an alternative, somewhat more powerful aspirin; they work similarly, have similar effects, and are used for similar purposes. Like aspirin, some people use it chronically and it can be given in acute events as well. It can “stack” with aspirin for a synergistic effect, or be used in its place for those who cannot tolerate aspirin.
Once again, the mechanism will sound familiar. One of the pathways that activates glycoprotein IIB/IIIA receptors requires the binding of adenosine diphosphate, or ADP. (ADP is more famous as the product of ATP once energy is released, but it has its fingers in a lot of cellular pies.) The thienopyridines block ADP binding and hence discourage platelet aggregation. Prasugrel (Effient) is another drug in this class.
Adverse effects generally involve bleeding diatheses.
More Drug Families: Stimulants and Depressants; Steroids and Antibiotics; ACE Inhibitors and ARBs
{kind=link}
Recent Comments